
Human Subjects in Research
University policy and federal law (45 CFR 4, 21 CFR Parts 50 and 56) require that all research involving human subjects, biospecimens, and/or identifiable private information , must be reviewed and approved by an Institutional Review Board (IRB) prior to the start of any research activities.
IRBNet
IRBNet is the protocol management system that offers secure, web-based collaboration tools to support the design, management, review and oversight of research involving human subjects.
IRBNET Submission Instructions
IRBNet Training
IRBNet is the protocol management system that offers secure, web-based collaboration tools to support the design, management, review and oversight of research involving human subjects. Detailed training on how to register as a new user and how to use IRBNet for project submissions is available by contacting hsrb-research@udel.edu.
To Register as an IRBNet user and log in for the first time
- Go to http://www.irbnet.org
- In upper right-hand corner of the screen, under the login boxes, click on New User Registration and complete the registration (fill in your first and last name, create a user name and a password), then click Continue. On the next screen about Terms of Use, click Accept.
- In the box Search for Organization, type University of Delaware (make sure Research Institutions is selected) and then click the Search button. In the search results, select University of Delaware, Newark, DE, and click Continue. Complete the required fields and click Continue (Red * indicates required fields). Verify that all information is correct and click the Register button affiliated with the University of Delaware IRB. At the Registration is Complete screen, click Continue.
- Next, you will receive an email from IRBNet with the subject Activation Required.
- Click on (or copy and paste) the IRBNet link in the your email message to return to the IRBNet site. Login with your IRBNet username and password to authenticate your registration.
To submit a NEW Project on IRBNet
- Go to http://www.irbnet.org and log in.
- In the blue menu bar on the left-hand side of the screen, choose Forms and Templates.
- In the Library pull down menu, select University of Delaware IRB – Documents for Researchers.
- From the list of documents in the library, download the University of Delaware Application Instructions if you are unfamiliar with the requirements or process.
- Download the New project protocol form and the applicable Informed Consent Form Template (Biomedical or Social-Behavioral-Educational) and save to your computer.
- Fill out the Protocol form, the Informed Consent and any other forms that may apply (e.g., Assent Form, Prisoners in Research Form, HIPAA authorization, etc.).
When you have all your study documents ready to submit:
- Log back into IRBNet.
- In blue menu bar on left-hand side of the page, choose Create New Project.
- Fill out the online information, including title, name, PI, etc. (Red * indicates required field) and click Continue.
- Click on the Add New Document button to upload your protocol form, consent form, surveys and questionnaires.
- Choose Sign this Package from the blue menu bar on the left-hand side of the screen to electronically sign the submission.
- Choose Submit this Package from the same blue menu bar to submit the Project for IRB review.
- Upon submission, you will receive an electronic notification via IRBNet.
To submit an Amendment, Continuing Review or Adverse Event form on IRBNet
- Go to http://www.irbnet.org and log in.
- In the blue menu bar on the left-hand side of the screen, choose Forms and Templates.
- From the Library pull down menu, select University of Delaware IRB – Documents for Researchers from the Library menu.
- Download to your computer and fill out the relevant form.
When you have your documents ready to submit:
- Log back into IRBNet.
- In the blue menu bar on the left-hand side of the screen, choose My Projects.
- Click on the Project Title to select the Project for which you are making the additional submission.
- Click on the Project History button on the left menu, then click on Create New Package box.
- In the blue menu bar on the left-hand side of the screen – click on Designer.
- Click on the Add New Document box to upload your documents.
- Choose Sign this Package from the blue menu bar on the left-hand side of the screen to electronically sign the submission.
- Choose Submit this Package from the same blue menu bar to submit the Project for IRB review.
- Upon submission, you will receive an electronic notification on IRBNet.
Educational Activities that ARE Human Subjects Research: If an instructor determines that there is a possibility that a student’s proposed research project may result in a formal presentation or publication, he/she should recommend that the student submit the project for IRB review before beginning the study.
Educational Activities that ARE NOT Human Subjects Research: All human subjects research requires prior institutional approval, but not all data gathering by students constitutes human subjects research. The definition of research below establishes that an activity must be designed with the intent to develop or contribute to “generalizable knowledge.” Classroom activities designed to teach research techniques or allow students to practice those techniques are not considered research with human subjects and are not required to be submitted to the UD IRB for review.
Simulations of human experimentation and course-assigned data collection do not constitute human subjects research if all of the following conditions are true:
- activities are designed for educational purposes only;
- the data will not be generalized outside the classroom (reporting of data within the class is acceptable because the activities were performed solely for teaching purposes);
- the data will not result in a master’s thesis or doctoral dissertation; and
- the student volunteers or other participants are clearly informed that the activities are an instructional exercise, and not actual research.
Human Subjects Protection (HSP)
Good Clinical Practice (GCP)
Definitions
Clinical Trial
Educational Activities
Educational Activities that ARE NOT Human Subjects Research: All human subjects research requires prior institutional approval, but not all data gathering by students constitutes human subjects research. The definition of research below establishes that an activity must be designed with the intent to develop or contribute to “generalizable knowledge.” Classroom activities designed to teach research techniques or allow students to practice those techniques are not considered research with human subjects.
Simulations of human experimentation and course-assigned data collection do not constitute human subjects research if the activities are designed for educational purposes only; and
- the data will not be generalized outside the classroom (reporting of data within the class is acceptable because the activities were performed solely for teaching purposes); and
- the data will not result in a master’s thesis, doctoral dissertation, poster session, abstract or other publication or presentation; and
- the student volunteers or other participants are clearly informed that the activities are an instructional exercise and not actual research.
Human subject
- Obtains information or biospecimens through intervention or interaction with the individual and uses, studies, or analyzes the information or biospecimens, or
- Obtains, uses, studies, analyzes, or generates identifiable private information or identifiable biospecimens.
Intervention
Interaction
Minimal Risk
Private information
Identifiable private information is private information for which the identity of the subject is or may readily be ascertained by the investigator or associated with the information.
An identifiable biospecimen is a biospecimen for which the identity of the subject is or may readily be ascertained by the investigator or associated with the biospecimen.
Research
HIPAA
How the Rule Works:
- By obtaining individual authorization: An Authorization is basically an individual’s written permission or consent to use his or her PHI for research purposes. HIPAA requires that an Authorization be written in plain language and contain certain “core” elements. Research authorizations may be combined with an informed consent form or set forth in a separate Authorization document. See forms and templates in IRBNet for further guidance on what to include in a HIPAA Authorization for research.
- By obtaining IRB waiver or alteration of the authorization requirement: The following three criteria must be satisfied for an IRB or Privacy Board to approve a waiver of authorization under the Privacy Rule:
- The use or disclosure of protected health information involves no more than a minimal risk to the privacy of individuals, based on, at least, the presence of the following elements:
- an adequate plan to protect the identifiers from improper use and disclosure;
- an adequate plan to destroy the identifiers at the earliest opportunity consistent with conduct of the research, unless there is a health or research justification for retaining the identifiers or such retention is otherwise required by law; and
- adequate written assurances that the protected health information will not be reused or disclosed to any other person or entity, except as permitted by this subpart;
- The research could not practicably be conducted without the waiver or alteration; and
- The research could not practicably be conducted without access to and use of the protected health information.
- The use or disclosure of protected health information involves no more than a minimal risk to the privacy of individuals, based on, at least, the presence of the following elements:
- By using de-identified information: Health information that has been “de-identified” in a manner required by HIPAA is not considered PHI and may be used or disclosed for research purposes without individual authorization. De-identification can be done by removal of all 18 elements that could be used to identify an individual and/or the individual’s relatives as described in the Privacy Rule. Alternatively, de-identification may be established by the use of statistical methods.
- By using limited data sets with a data use agreement: A limited data set is described as health information that excludes certain listed direct identifiers but that may include city, state, ZIP Code, elements of date and other numbers, characteristics or codes not listed as direct identifiers. It is the responsibility of the researcher and the party releasing the PHI to have in place and maintain a copy of a data use agreement that meets HIPAA requirements.
- By using only decedents’ information, with certain assurances
- By using PHI for purposes preparatory to research, with certain assurances and with no removal of any PHI from the covered entity (physically or electronically).
Related Policies / Procedures
- Template: Documentation Consent Process Form and Log
- Procedure: Administrative Review
- Procedure: Delay on the Updated Common Rule Effective Date
- Procedure: Exemptions
- Procedure: Expedited Review
- Procedure: Full Board Review
- Procedure: IRB Review Types
- Procedure: Schematic Representations of a Study Examples
- Policy: Case Study vs. Research UD Guidance
- Policy: HIPAA Hybrid Statement
- Policy: Human Subjects in Research and Research-Related Activities
- Policy: Ratner Prestia Guide to Intellectual Property
- Policy: Research vs. Quality Assurance / Improvement Guidance
- Form: Exempt Determination Tool
- Form: IRBNet New User Registration
IRB Overview
University of Delaware Institutional Review Board
The University of Delaware has one IRB registered with DHHS (IORG #0000279). UD IRB membership is in accordance with the applicable regulatory requirements in 45 CFR 46.107 and 21 CFR 56.107. IRB membership includes diverse backgrounds and expertise to promote adequate review of research commonly conducted by UD researchers. Membership shall include both scientists and non-scientists, as well as outside community members. Members from the UD faculty and staff are to be nominated by their respective dean, department chair or supervisor, in concurrence with the IRB Chair and the UD Vice President for Research, Scholarship and Innovation. Outside community members (not affiliated with the UD community) will be nominated by the IRB Chair. The UD IRB members, including the IRB Chair, are appointed by the UD Vice President for Research, Scholarship and Innovation.
Members shall serve for terms of three years, renewable with concurrence of their respective dean, department chair or supervisor, the IRB Chair and the UD Vice President for Research, Scholarship and Innovation. All members will complete human subjects protections and IRB training prior to starting each term. Members are expected to attend regularly scheduled IRB meetings.
IRB members may have designated alternate(s) members. An alternate is an individual appointed to the IRB to serve in the same capacity as the specific IRB member(s) for whom the alternate is named. Alternate members must meet the same training requirements as the full member for which they serve.
The UD IRB Office supports the functions of the IRB and its Chair, Shannon Lennon, Ph.D. The IRB can be contacted at:
University of Delaware IRB
STAR Tower, 10th Floor (Research Office)
Phone: 302-831-2137
Email: hsrb-research@udel.edu
UD IRB meetings are held monthly. Generally, meetings will be at noon on the third Wednesday of each month. Meeting dates and times are posted on the Research Office calendar of events.
For convened IRB meetings the required quorum is defined as more than half of the current full membership. At least one member whose primary concerns are in non-scientific areas, and one member whose primary concerns are in scientific areas must be in attendance to satisfy quorum requirements. In addition, if research involving prisoners is to be reviewed, the member designated as the prisoner advocate must also attend.
More
IRB members must avoid potential conflicts of interest when conducting protocol review. A potential conflict of interest occurs when a reasonable outside observer might perceive the circumstances as creating an apparent conflict of interest. Examples of such potential conflicts include an IRB member having a close personal relationship with a researcher submitting a proposal, an IRB member serving as a researcher on the proposed project, or an IRB member serving as a consultant to the project. IRB members with potential conflict of interest must recuse themselves and not be present for the discussion, except as to provide information requested by the IRB, and vote on the research. Recused members will not be counted towards the quorum requirements in the review and voting of the research for which they recused.
Quorum will be determined and verified by the IRB office staff member(s) attending the meeting before the discussion and vote for each item reviewed. IRB decisions will be made based on the vote of the majority of the eligible members present at the meeting.
Participation in a convened meeting via tele or video conference is acceptable to meet quorum requirements provided the member(s) have received the materials to be reviewed prior to the meeting and can actively participate in the discussion.
Federalwide Assurance FWA
The University of Delaware has a Federalwide Assurance FWA (#00004379) on file with the Department of Health and Human Services (DHHS) Office for Human Research Protections (OHRP). Through this document the University commits itself to upholding the Code of Federal Regulations and the ethical principles of the Belmont Report for all research involving human subjects conducted by University faculty, staff and students. The UD Vice President for Research, Scholarship and Innovation is the Signatory Official of the UD FWA.
For questions please contact the UD IRB Office or (302) 831-2137
Exemptions
Exemptions Effective as of 1/21/2019
Research meeting the criteria described in the following categories can be deemed exempt from regulatory requirements imposed by the Common Rule. Exempt review determinations MUST be made by the IRB office and require the submission of a research protocol to the IRB.
CATEGORY
- Research, conducted in established or commonly accepted educational settings, that specifically involves normal educational practices that are not likely to adversely impact students’ opportunity to learn required educational content or the assessment of educators who provide instruction.This category would apply to most research on regular and special educational instructional strategies, and research on the effectiveness or the comparison among instructional techniques, curricula, or classroom management methods.
- Established or commonly accepted educational settings are settings where one would go in order to have an educational experience that is regularly offered, or that is commonly accepted in a specific culture or population.
- Normal educational practices are activities that could occur in the specific educational setting regardless of whether the research is conducted. This includes a variety of activities that normally occur in the classroom or that are considered “best practice”. Examples include established teaching methods (not considered to be experimental) or curriculum, and commonly accepted classroom management techniques that are planned and implemented by the classroom teacher.
- Educational tests, surveys, interviews, observations of public behavior: Research that only includes interactions involving educational tests (cognitive, diagnostic, aptitude, achievement), survey procedures, interview procedures or observation of public behavior (including visual or auditory recordings) if at least one of the following criteria is met:
- The information obtained is recorded by the investigator in such a manner that the identity of the human subjects cannot be readily ascertained, directly or through identifiers linked to the subjects.
- Any disclosure of the human subjects’ responses outside the research would not reasonably place the subjects at risk of criminal or civil liability or be damaging to the subjects’ financial standing, employability, educational advancement, or reputation.
- The information obtained is recorded by the investigator in such a manner that the identity of the human subjects can readily be ascertained, directly or through identifiers linked to the subjects, and an IRB conducts a limited IRB review to make the determination that, when appropriate, there are adequate provisions to protect the privacy of subjects and to maintain the confidentiality of data.
- Research involving children does not qualify for this exempt category if: (1) the research involves surveys, interviews, and/or observations of public behavior when the research team participates in the activities being observed, or (2) if Limited IRB Review is required.
- Observation of public behavior must not be influenced by the investigator and cannot involve an intervention (e.g., research involving observation of public behavior does not qualify for this exemption if the investigator intervenes with subjects by asking a question or posting a comment with on a public chat room with the intent to change the environment to observe behavior changes.
- Benign behavioral interventions. Research involving benign behavioral interventions in conjunction with the collection of information from an adult subject through verbal or written responses (including data entry) or audiovisual recording if the subject prospectively agrees to the intervention and information collection and at least one of the following criteria is met:
- The information obtained is recorded by the investigator in such a manner that the identity of the human subjects cannot readily be ascertained, directly or through identifiers linked to the subjects;
- Any disclosure of the human subjects’ responses outside the research would not reasonably place the subjects at risk of criminal or civil liability or be damaging to the subjects’ financial standing, employability, educational advancement, or reputation; or
- C. The information obtained is recorded by the investigator in such a manner that the identity of the human subjects can readily be ascertained, directly or through identifiers linked to the subjects, and an IRB conducts a limited IRB review to determine that the research (when appropriate) has adequate provisions to protect the privacy of subjects and to maintain the confidentiality of data.
Research involving children does not qualify for this exempt category.- Benign behavioral interventions are defined as brief in duration, harmless, painless, not physically invasive, not likely to have a significant adverse lasting impact on the subjects, and the researcher has no reason to think that the subjects will find the interventions/ interactions/observations to be offensive or embarrassing.
- Prospective agreement. Subjects must be asked to agree to participate in the research. This is not the same as the requirement for consent, or for documentation of consent. The request may be tailored to the nature of the specific study.
- Deception. If the research involves deceiving the subjects regarding the nature or purposes of the research, this exemption is not applicable unless the subjects authorizes the deception through a prospective agreement to participate in research in circumstances in which the subject is informed that he or she will be unaware of or misled regarding the nature or purposes of the research.
- Secondary research uses of identifiable private information or identifiable biospecimens for which consent is not required, if at least one of the following criteria is met:
- Publicly available. The identifiable private information or identifiable biospecimens are publicly available.
- Not identifiable as recorded. Information, which may include information about biospecimens, is recorded by the investigator in such a manner that the identity of the human subjects cannot readily be ascertained directly or through identifiers linked to the subjects, the investigator does not contact the subjects, and the investigator will not re-identify subjects.
- Use of PHI. The research involves only information collection and analysis involving the investigator’s use of identifiable health information when that use is regulated under HIPAA regulations, for the purposes of health care operations, research, or public health activities and purposes (as those purposes are described in the HIPAA regulations).
- Use of federally generated or collected information or biospecimens. The research is conducted by, or on behalf of, a federal department or agency using government-generated or government-collected information originally obtained for non- research activities, if the original collection and the secondary use of the information or biospecimens occurs in compliance with three specific federal statues meant to safeguard privacy.
- Secondary means re-using identifiable information and identifiable biospecimens that are collected from some other “primary” or “initial” activity; in other words, not for the purpose of the specific proposed study.
- “For which consent is not required” is interpreted as there are no federal or state laws that require subject consent for the proposed secondary use. During the original collection of the information or biospecimens, the individuals (if asked) agreed to secondary uses that were described in a manner consistent with the proposed research.
- Research and demonstration projects that are conducted or supported by a federal department or agency, or otherwise subject to the approval of department or agency heads (or the approval of the heads of bureaus or other subordinate agencies that have been delegated authority to conduct the research and demonstration projects), and that are designed to study, evaluate, improve, or otherwise examine public benefit or service programs, including procedures for obtaining benefits or services under those programs, possible changes in or alternatives to those programs or procedures, or possible changes in methods or levels of payment for benefits or services under those programs.All of the following criteria must be met under this exemption:
- The program under study delivers a public benefit or service.
- The project must be conducted pursuant to specific federal statutory authority.
- There must be no statutory requirement that the project be reviewed by an IRB.
- The project does not involve significant physical invasions or intrusions upon the privacy of participants.
- The project does not involve significant physical invasions or intrusions upon the privacy of participants.
Requirement for the federal department or agency conducting or supporting the project. The federal department or agency conducting or supporting the project must establish, on a publicly accessible federal Web site or in such other manner as the department or agency head may determine, a list of the research and demonstration projects the federal department or agency conducts or supports under this exempt category. The department or agency head can determine what sort of information will be included on this list and maintains its oversight. The project must be published on the list before the researcher can begin the project; however, exempt status can be granted before the publication occurs. - Taste and food quality evaluation and consumer acceptance studies:
- if wholesome foods without additives are consumed, or
- if a food is consumed that contains a food ingredient at or below the level and for a use found to be safe, or agricultural chemical or
- environmental contaminant at or below the level found to be safe, by the Food and Drug Administration or approved by the Environmental Protection Agency or the Food Safety and Inspection Service of the Department of Agriculture.
The UD IRB has not implemented exempt determinations under categories 7 and 8 at this time. Brief descriptions are provided here for information purposes
- Storage or maintenance for secondary research for which broad consent is required. Storage or maintenance of identifiable private information or identifiable biospecimens for potential secondary research use if an IRB conducts a limited IRB review and makes all of the following determinations:
- Broad consent for storage, maintenance, and secondary research use of identifiable private information or identifiable biospecimens is obtained in accordance with the requirements prescribed by the Rule
- Broad consent is appropriately documented or waiver of documentation is appropriate.
- If there is a change made for research purposes in the way the identifiable private information or identifiable biospecimens are stored or maintained, there are adequate provisions to protect the privacy of subjects and to maintain the confidentiality of the data.
- Secondary research for which broad consent is required. Research involving the use of identifiable private information or identifiable biospecimens for secondary research use, if all of the following criteria are met:
- Broad consent for the storage, maintenance, and secondary research use of the identifiable private information or biospecimens was obtained in accordance with all of the requirements described for exempt category 7.
- Documentation of informed consent or waiver of documentation of consent was obtained.
- An IRB conducts a limited IRB review and makes the following determinations:
- When appropriate, there are adequate provisions to protect the privacy of subjects and to maintain the confidentiality of the data.
- The research to be conducted is within the scope of the broad consent provided by the subjects. The investigator does not include returning individual research results to subjects as part of the study plan. (Note: this provision does not prevent an investigator from abiding by any legal requirements to return individual research results.)
Expedited Review
Expedited review procedures may be used for certain research activities described in the federal regulations. Expedited reviews are done by one or more experienced reviewers designated by the chairperson from among members of the IRB. Projects approved by the expedited review process are subject to the same regulatory requirements as those approved on a full board review and must be periodically reviewed by continuing review before the expiration date set on approval (no longer than one year since approval). Informed consent forms associated with projects reviewed by expedited review will be stamped by the IRB with the approval and expiration dates. IRB-stamped documents are posted in IRBNet after approval and must be used when obtaining the informed consent of research participants.
Protocols eligible for expedited review are evaluated on a rolling basis as they are submitted to IRBNet. Review times for expedited reviews vary depending on the total IRB submissions load at any given time and may take, on average, about three weeks from the time of complete submission.
- Clinical studies of drugs and medical devices only when condition (a) or (b) is met.
(a) Research on drugs for which an investigational new drug application (21 CFR Part 312) is not required. (Note: Research on marketed drugs that significantly increases the risks or decreases the acceptability of the risks associated with the use of the product is not eligible for expedited review.)
(b) Research on medical devices for which (i) an investigational device exemption application (21 CFR Part 812) is not required; or (ii) the medical device is cleared/approved for marketing and the medical device is being used in accordance with its cleared/approved labeling. - Collection of blood samples by finger stick, heel stick, ear stick or venipuncture as follows:
(a) from healthy, nonpregnant adults who weigh at least 110 pounds. For these subjects, the amounts drawn may not exceed 550 ml in an eight-week period and collection may not occur more frequently than two times per week; or
(b) from other adults and children, considering the age, weight and health of the subjects, the collection procedure, the amount of blood to be collected and the frequency with which it will be collected. For these subjects, the amount drawn may not exceed the lesser of 50 ml or 3 ml per kg in an eight-week period and collection may not occur more frequently than two times per week. - Prospective collection of biological specimens for research purposes by noninvasive means. Examples:
(a) hair and nail clippings in a nondisfiguring manner;
(b) deciduous teeth at time of exfoliation or if routine patient care indicates a need for extraction;
(c) permanent teeth if routine patient care indicates a need for extraction;
(d) excreta and external secretions (including sweat);
(e) uncannulated saliva collected either in an unstimulated fashion or stimulated by chewing gumbase or wax or by applying a dilute citric solution to the tongue;
(f) placenta removed at delivery;
(g) amniotic fluid obtained at the time of rupture of the membrane prior to or during labor;
(h) supra- and subgingival dental plaque and calculus, provided the collection procedure is not more invasive than routine prophylactic scaling of the teeth and the process is accomplished in accordance with accepted prophylactic techniques;
(i) mucosal and skin cells collected by buccal scraping or swab, skin swab or mouth washings;
(j) sputum collected after saline mist nebulization. - Collection of data through noninvasive procedures (not involving general anesthesia or sedation) routinely employed in clinical practice, excluding procedures involving x-rays or microwaves. Where medical devices are employed, they must be cleared/approved for marketing. (Studies intended to evaluate the safety and effectiveness of the medical device are not generally eligible for expedited review, including studies of cleared medical devices for new indications.) Examples:
(a) physical sensors that are applied either to the surface of the body or at a distance and do not involve input of significant amounts of energy into the subject or an invasion of the subject’s privacy;
(b) weighing or testing sensory acuity;
(c) magnetic resonance imaging;
(d) electrocardiography, electroencephalography, thermography, detection of naturally occurring radioactivity, electroretinography, ultrasound, diagnostic infrared imaging, doppler blood flow and echocardiography;
(e) moderate exercise, muscular strength testing, body composition assessment and flexibility testing where appropriate given the age, weight and health of the individual. - Research involving materials (data, documents, records or specimens) that have been collected or will be collected solely for nonresearch purposes (such as medical treatment or diagnosis).
(NOTE: Some research in this category may be exempt from the HHS regulations for the protection of human subjects. 45 CFR 46.101(b)(4). This listing refers only to research that is not exempt.) - Collection of data from voice, video, digital or image recordings made for research purposes.
- Research on individual or group characteristics or behavior (including, but not limited to, research on perception, cognition, motivation, identity, language, communication, cultural beliefs or practices, and social behavior) or research employing survey, interview, oral history, focus group, program evaluation, human factors evaluation or quality assurance methodologies.
(NOTE: Some research in this category may be exempt from the HHS regulations for the protection of human subjects. 45 CFR 46.101(b)(2) and (b)(3) . This listing refers only to research that is not exempt.) - Continuing review of research previously approved by the convened IRB as follows:
(a) where (i) the research is permanently closed to the enrollment of new subjects; (ii) all subjects have completed all research-related interventions; and (iii) the research remains active only for long-term follow-up of subjects; or
(b) where no subjects have been enrolled and no additional risks have been identified; or
(c) where the remaining research activities are limited to data analysis. - Continuing review of research not conducted under an investigational new drug application or investigational device exemption where categories two (2) through eight (8) do not apply but the IRB has determined and documented at a convened meeting that the research involves no greater than minimal risk and no additional risks have been identified.
Full Board Review
Review of non-exempt research that does not qualify for expedited review or that may present more than minimal risk to the subjects must be reviewed at a convened meeting of the University’s IRB. At the University of Delaware (UD) the IRB meets once every month and investigators proposing new protocols are usually invited to attend the IRB meeting to present their research and address any questions the Board members may have. Students having protocols reviewed at a convened meeting and presenting to the IRB must be accompanied by their faculty advisor.
The meeting dates for the IRB as well as the submission deadline for protocols to be considered for full board review at each month’s meeting are set well in advance and can be found in the Research Office Calendar of Events. The number of protocols to be reviewed at each meeting may be limited due the constraints of time and the complexity of other items on the agenda. Protocols will be accepted for review in the order received. If the board is not able to review a protocol in a particular month, it will be given priority for review in the following month.
Investigators submitting new projects are normally invited to attend the IRB meeting to present their project to the Board and answer any questions or concerns IRB Members may have. The results from the meeting are later communicated to the PI and any conditions the IRB may have set, before approval can be effective, will be addressed then. If a student-led new project is to be reviewed by the full board the academic advisor responsible for the student and project must accompany the student to the meeting.
Projects approved at a convened full board meeting must be periodically reviewed by continuing review before the expiration date set on approval and no less than once a year. Informed consent forms associated with projects reviewed at a full board will be stamped by the IRB with the approval and expiration dates. IRB-stamped documents are posted in IRBNet after approval and must be used when obtaining the informed consent of research participants.
Admin Review
In addition to the types of review listed above the IRB may provide administrative reviews when appropriate. Administrative reviews are used in cases in which the University of Delaware needs record of research with human subjects in which UD is engaged but for which review and approval are not issued by the UD IRB. This is the case, for example, when UD investigators are part of a research team performing research at another institution. Depending on the nature of the research and the collaboration arrangement, UD IRB may rely on the review and approval from the other institution’s IRB. UD keeps record of that reliance via an administrative review.
All UD investigators engaged in collaborative research with human subjects must consult with the UD IRB office and submit that proposed effort to the UD IRB regardless of other IRB reviews being sought after.
New Projects
Here is an overview of the Life Cycle of an IRB Protocol.
All research with human subjects performed by University of Delaware researchers must be reviewed and approved by the UD IRB. Submissions to the University of Delaware IRB must be made using the IRBNet protocol management system (www.irbnet.org). Step-by- step instructions on how to navigate IRBNet and further training on how to use the system can be seen above.
Collaborative Research that involves UD researchers must be submitted to the UD IRB for review even if the project is to be reviewed and approved by another IRB. The submission package must include the approval letter, the protocol form submitted to the other institution as well as the UD protocol form clearly describing the specific role and data access the UD personnel will have in the project.
Student-led projects to be reviewed by the IRB must be shared and signed off in IRBNet by the faculty advisor responsible for the student and project. Student projects will not be reviewed until signed by the faculty advisor in IRBNet.
Any research involving human subjects, bio specimens and/or tissue samples from humans, and/or private identifiable data must be reviewed by an Institutional Review Board (IRB). Submissions for review must be done using IRBNet and all new project application packages must include the protocol form properly filled out and, when applicable, the informed consent document(s), and any other relevant materials (e.g., advertisements, research instruments, surveys, questionnaires, etc.). Forms and templates are available in IRBNet under the “Forms and Templates” tab in the left-hand side menu options.
Once submitted the IRB office will determine the type of review needed. If the project is deemed to be exempt or suitable for an expedited review, the review will be performed on an ongoing basis as the submission is received. Processing times will vary depending on the total workload of the IRB at a given time. When a new project is determined to require full board review it will be added to the next available IRB meeting agenda as it is received. The IRB at UD meets once a month and deadlines for submissions to be considered for review are posted in the research calendar. After review the IRB office will communicate with the principal investigator and request for any clarifications or edits needed to be completed before approval can be issued.
Once a review has been completed, decision letters are uploaded in IRBNet and are always accessible to the investigator(s). Informed consent documents from projects approved via an expedited or full board review will be stamped with the IRB approval and expiration date and also uploaded in IRBNet. Informed consent must be obtained using the stamped version of the approved documents, (i.e., participants signatures need to be collected in a copy of the stamped informed consent). No IRB stamp is added to informed consent documents associated to projects deemed to be exempt.
For questions, send email to: hsrb-research@udel.edu
Amendments
If ANY changes need to be made to an IRB approved protocol, investigators must seek IRB approval of those changes prior to their implementation. Examples of changes that would require the submission of an approval are: changes to the research team members, modifications in the recruitment strategies and/or advertisement materials, any changes to the research instruments already approved, elimination of previously approved procedures and/or data collection measures, new approaches to data processing or storage, edits to the approved informed consent language, etc. Amendments are reviewed as they are submitted and may take on average up to two-weeks to be completed.
Amendment submission packages must contain:
- the amendment form properly filled out,
- the tracked changes and clean versions of any documents affected by the amendment (e.g., edits to the previously approved protocol form and informed consent document, advertisement materials, etc.) Tracked changes version of any edited document must show all the markups including what is being added and/or removed.
If the amendment does affect the informed consent document, a clean version (changes accepted) needs to be submitted so a newly IRB-stamped informed consent can be issued upon approval. The approval of an amendment normally has no effect on the previously set expiration date.
Continuing Review
Protocols approved by the IRB, whether under expedited or full board review, are required to undergo continuing review at least once per year, on or before the expiration date set at the time of approval for as long as recruitment and data collection is to take place, and/or private identifiable data is kept. In some cases, the IRB may require more frequent continuing reviews. Expiration reminders are automatically sent from IRBNet to the Principal Investigator (PI) and all others with whom the project has been shared, and granted full access to, in IRBNet. Reminders are sent 60 and 30 days before the project is set to expire. In addition, an expiration alert is sent on the expiration day if no approval has been secured before that day. Applications for continuing review must be submitted with enough time to allow for IRB review prior to the expiration date. In order to maintain the expiration anniversary date, continuing review applications are reviewed no earlier than 30 days before the expiration date (because of this, a 30-day expiration notice will always be received even when the application for review has been submitted early). Once a continuing review is approved a new one-year approval period starts and informed consent documents are stamped with the new approval and expiration dates.
Unless eligible for expedited continuing review, protocols originally reviewed and approved by the convened IRB meeting (full board) must undergo a full board continuing review. IRB meeting dates and deadlines must be considered when submitting a continuing application to be reviewed at a full board as to avoid lapses in IRB approval. Continuing review submissions must contain the continuing review form properly filled in and all other current documents relevant to the project (at a minimum the most current previously approved protocol form and a clean version of the informed consent). If any changes are added at the time of continuing review, the tracked changes version of the protocol form and informed consent document(s) need to be added to the continuing review application.
Lapses in IRB Approval: It is the responsibility of the investigators to provide in a timely manner the information needed by the IRB to perform its continuing review functions. When continuing review of a research project is not completed prior to the end of the approval period specified, the IRB approval will expire. All research activities involving human subjects (i.e., participant recruitment and/or data collection) must stop after IRB approval expires and cannot be restarted until approval has been secured. Expired projects for which the IRB office has not been notified of the intent for continuation will be closed. Once closed, a project cannot be re-opened and will have to be resubmitted as ‘new’ for review and approval before any research related activities can be re-initiated.
Problems
Investigators must report to the IRB Office any instances of unanticipated problems (UP), and/or adverse events (AE) related to the research within three days of the incident.
Unanticipated problems, in general, include any incident, experience or outcome that meets the following criteria:
- unexpected (in terms of nature, severity or frequency) given (a) the research procedures that are described in the protocol-related documents, such as the IRB-approved research protocol and informed consent document; and (b) the characteristics of the subject population being studied;
- related or possibly related to participation in the research (in this guidance document, possibly related means there is a reasonable possibility that the incident, experience or outcome may have been caused by the procedures involved in the research);
- suggests that the research places subjects or others at a greater risk of harm (including physical, psychological, economic or social harm) than was previously known or recognized.
Adverse Events
An adverse event is any untoward or unfavorable medical occurrence in a human subject, including any abnormal sign (e.g., abnormal physical exam or laboratory finding), symptom or disease, temporally associated with the subject’s participation in the research, whether or not considered related to the subject’s participation in the research. Adverse events encompass both physical and psychological harms. They occur most commonly in the context of biomedical research, although on occasion, they can occur in the context of social and behavioral research.
Unanticipated problems and adverse events must be reported to the IRB by submitting the appropriate form via IRBNet. Depending on the nature and of the event reported and on a case by case basis the Director of Research Compliance and the IRB Chair will decide if review by the convened IRB of the submission is needed and when a for cause audit of the research project may be required.
Audits
Auditing will be performed routinely by the IRB office staff to ascertain general compliance with the protection of human subjects requirements and IRB-approved protocol and procedures. In addition, for cause audits will be conducted as deemed appropriate by the Director of Research Compliance, and/or the IRB Chair, to investigate reports of unanticipated problems, adverse events and/or non-compliance.
- Routine Audits: Projects are selected randomly among those active and approved via expedited and convened IRB review. The principal investigator responsible for the project is contacted to schedule a visit from the IRB office staff member conducting the audit. Prior to the scheduled visit the investigator is sent an outline of the review process. A report is generated after the audit and sent to the investigator for review. Non-compliance and other findings resulting from a routine audit are shared with the IRB. If a non-compliance finding is determined by the IRB to be serious or continuing it will be further investigated and reported to the UD Signatory Official.
- For cause Audit: Adverse events and/or unanticipated problems involving risk to participants reported to the IRB office may be followed up with an investigation. For cause audits may also be performed to investigate non-compliance with IRB-approved protocols. Depending on the nature of the investigation, for cause audits may be scheduled or unannounced. Reports from a for cause audit will be reviewed by the IRB. Depending on the outcome of a for cause audit and the IRB review of the report, an incident may have to be reported to the UD Signatory Official. The UD Signatory Official has the authority to decide if, as per the stipulated guidance, further reporting to OHRP and the funding agency is needed.
Closure
Protocols approved by the IRB under an expedited or full board review must receive continuing review at least once per year until closed. A project can be closed when all the following conditions are true:
- Enrollment and all data collection for the study have been completed; private identifiable data is no longer being stored, accessed or worked on
- the link (access code), if any, between the research data and the identifiers has been destroyed. More details on data storage and retention can be found in the data management section below.
A closure submission should be done as soon as the conditions above are met by creating a new submission package in IRBNet and including the closure form. If no continuing review application nor closure request is submitted by the investigator, projects will be closed by the IRB Office upon expiration. Once a study has been closed it cannot be re-opened. Any future work related to the study (e.g., long- term follow up with participants, additional enrollment and data collection, etc.) will require for a new project to be reviewed and approved by the IRB before the work can start.
Informed Consent
Informed Consent Process
and Documentation
DownloadInformed Consent Process and Documentation
Informed consent is a process and involves providing a potential subject with adequate information about the research to allow for an informed decision about the subject’s voluntary participation in a research study. Informed consent is a process. Documenting informed consent occurs after explaining the research, addressing any questions, and assessing participant comprehension.
Purpose
To provide guidance about the informed consent process and documentation of consent procedures in research with human subjects:
- Suggested best practices in obtaining and documenting informed consent from prospective research participants
- Description of Regulatory and IRB requirements related to documentation of informed consent
- Considerations when informed consent is to be sought remotely.
- Requirements for electronic documentation of informed consent.
- Documentation of combined informed consent and HIPAA authorization documents.
Documentation of Informed Consent Regulatory Requirements
- Federal Policy requires that legally effective informed consent to participate in research must be obtained and documented by investigators prior to involvement of participants in non-exempt human subjects research, unless a waiver or alteration of informed consent is approved by an IRB.
- Informed consent is a process and involves providing a potential subject with adequate information about the research to allow for an informed decision about the subject’s voluntary participation in a research study.
- Consent must be sought under circumstances that allow the prospective subject, or their legally authorized representative, the opportunity to discuss and consider whether to participate.
- Language used should be understandable to the subject, organized and presented in a way that facilitates comprehension.
- All consent procedures (remote or otherwise) must be described in the Research Protocol. Consider whether the research plan can benefit from outlining both remote and in-person consent procedures to maximize flexibility.
- Documentation of consent provides a record that the consent process took place. The requirement to document informed consent prescribes the use of a written form approved by the IRB and signed (handwritten or electronical signatures are acceptable) by the subject or the subject’s legally authorized representative (LAR).
- Informed consent must include several basic elements (i.e., statement that study includes research, risks, benefits, alternatives, confidentiality, compensation, contact information, voluntary participation, statement about future use of information and/or biospecimens) and may also include other information as appropriate (unknown risks, termination of participation, costs to subjects, consequences of withdrawal, new findings, etc.). Any alterations to the required elements of informed must be approved by the IRB.
- The most current UD consent form templates posted in IRBNet should be used in the development of informed consent documents. Researchers must check for updates of those templates every time they submit a new study for review to the IRB.
- In addition to the informed consent forms signed by the research participants (or their legally authorized representative or parent(s) when approved), researchers should keep record of the consent process having been completed. That documentation can be a cover page added to each participant record, or a consent process log documenting every subject consent process. Suggested templates for the consent process documentation forms are posted under IRBNet’s Forms and Templates section.
Waiver or alteration of Documentation of Informed Consent
- If written documentation of consent is not required per the federal regulations, researchers can request a waiver of documentation of consent and, if applicable and approved by the IRB, obtain verbal consent remotely via phone or teleconference platforms such as Zoom.
- A waiver or alteration of the requirement to document is not a waiver of the requirement to obtain informed consent. An IRB may waive the requirement to obtain the subjects’ signature on the informed consent for some or all subjects if it finds any of the following:
- The principal risks are those associated with a breach of confidentiality concerning the subject’s participation in the research, and the consent document is the only record linking the subject with the research. (e.g., collection of anonymous sensitive information).
- Under this criterion for waiver of documentation, each subject needs to be asked if they want documentation linking them with the research, and their wishes will govern. Informed consent document should include the signature line for subjects to sign as an option, if they wish to do so.
- The research presents minimal risk of harm and does not involve any procedures for which written consent would normally be required outside of the research context. (e.g., research that only involves non-sensitive surveys and questionnaires procedures could request a waiver to obtained signed informed consent documents).
- The subjects are members of a distinct cultural group or community in which signing forms is not the norm provided the study is minimal risk and there is an appropriate alternative mechanism for documenting that informed consent was obtained.
- The principal risks are those associated with a breach of confidentiality concerning the subject’s participation in the research, and the consent document is the only record linking the subject with the research. (e.g., collection of anonymous sensitive information).
- In cases in which the signature requirement is waived or alteration approved, it is considered best practice for the investigator and study team to record that the consent process has occurred via a documentation of consent memo or note to file. The proposed plan for consent process documentation must be clearly described in the protocol form.
- Exempt human subjects research does not require written documentation of consent, nor a request for a waiver of written documentation. An informed consent process is still recommended, and the abbreviated Informed Consent Template for Exempt research is available in IRBNet’s Forms and Templates section.
Remote Informed Consent Considerations
- In cases in which investigators and research participants will not be in the same physical location the informed consent process can be done remotely ensuring that:
- The remote consent procedures allow participants to experience a consent process as close to what it would be like in-person as possible (e.g., conversation with investigators allowing prospective participants to ask any questions they may have prior to giving informed consent). The participant should have ample time and opportunity to review the consent form in advance, and then discuss it and ask any questions together with the investigator
- For an online survey where no direct interaction with the participant will occur, it is permissible to construct the survey with an embedded consent form at the beginning whereby completion of the survey indicates the participant’s consent.
- The IRB-approved and stamped consent form is used.
- The IRB-approved Research Protocol includes an accurate description of the entire consent process
- The physical location of the investigator and participant can be any place convenient to them but must provide adequate space for privacy and confidentiality. Remote conference tools can be used for online or on the phone conversations. Video conferencing (e.g., Zoom) is allowable. Regardless of the environment, the participant must be informed in advance if the consent process will be audio and/or video recorded.
- The remote consent procedures allow participants to experience a consent process as close to what it would be like in-person as possible (e.g., conversation with investigators allowing prospective participants to ask any questions they may have prior to giving informed consent). The participant should have ample time and opportunity to review the consent form in advance, and then discuss it and ask any questions together with the investigator
- If written documentation is obtained remotely, the investigator must provide the person signing the consent form with a copy of the consent document unless this requirement is waived by the IRB. Copies can be sent via secure email, standard mail or document delivery service, fax, hyperlink for download on a secure website, etc. Enough copies should be provided that, if the participant is expected to return a hard copy, a second copy remains in their possession.
- If written documentation of consent (i.e., signature) is required, the participant can sign the consent during the remote consent process, as witnessed by the investigator, and it can be returned via one of the methods noted below.
- The regulations consider written documentation of consent to include electronic format. Acceptable options to obtain electronic signatures remotely are:
If using a digital (e-consent) form:
- Consent document(s) can be provided on a secure online platform with an electronic signature collected at the end (e.g. REDCap, DocuSign, Google Forms, online survey using the Signature option in Qualtrics ). Should this method be used, it is preferable to use a platform that is easy to navigate, allows the participant to stop, save, and/or move forward and backward within the form.
- Electronic signature – a computer data compilation of any symbol(s) executed, adopted, or authorized by an individual to be a legally binding equivalent of the individual’s handwritten signature. For studies under FDA oversight, electronic signatures must comply with 21 CFR 11.5 & 11.7 signature requirements:
- The printed name, or handwritten signature executed to the electronic document, of the signee;
- Date and time when the signature was executed;
- Meaning (i.e., consent); and
- Linked to their respective electronic records to ensure that it cannot be excised, copied, or otherwise transferred (i.e., tampered with).
- Electronic signature – a computer data compilation of any symbol(s) executed, adopted, or authorized by an individual to be a legally binding equivalent of the individual’s handwritten signature. For studies under FDA oversight, electronic signatures must comply with 21 CFR 11.5 & 11.7 signature requirements:
- Consent language must be exactly as approved by the IRB in the most current version. When possible, the exact IRB approval stamp needs to be incorporated into the electronic version shared with the participants. If the electronic platform does not support the use of the IRB-stamped version of the informed consent, the e-version must include the IRB approval date and document version (Package IRBNet ID#)
If using a paper consent form sent to a participant in advance of the consent discussion:
- Collect documentation as scanned copy or picture of the signed form sent back to investigators via secured email/file transfer.
- Collect a signed hard copy via fax or mail. Postage and mailing envelop/address should be provided by the investigator. In this case there should not be a place for the researcher to sign and date on the form itself. It is recommended to use a consent or enrollment log to capture this information instead.
Documentation of Combined Informed Consent and HIPAA Authorization for research
- The HIPAA Rules regulate how protected health information may be obtained and used for research purposes. This is true whether the PHI is completely identifiable or partially de-identified in a limited data set.
- To access and use PHI for research purposes appropriate HIPAA documentation must be obtained, including either:
- Individual patient authorization; or
- Approved waiver of authorization from an IRB or Privacy Board
- HIPAA authorizations for research purposes can be obtained on a separate document or combined with the Informed Consent document.
- The UD IRB provides templates containing the required elements to be included in a HIPAA authorization. Signature of the individual and date are required as documentation of HIPAA authorization.
- A waiver or alteration of HIPAA authorization can only be approved by an IRB or Privacy Board when the following criteria are met:
- The use or disclosure of PHI involves no more than a minimal risk to the privacy of individuals, based on, at least, the presence of the following elements:
- An adequate plan to protect the identifiers from improper use and disclosure;
- An adequate plan to destroy the identifiers at the earliest opportunity consistent with conduct of research, unless there is health or research justification for retaining the identifiers or such retention is otherwise required by law; and
- Adequate written assurances that the PHI will not be reused or disclosed except as required by law, for authorized oversight of the research study, or for other research for which the use or disclosure of PHI would be permitted by the Privacy Rule;
- The research could not practicably be conducted without the waiver or alteration; and
- The research could not practicably be conducted without access to and use of the PHI.
- The use or disclosure of PHI involves no more than a minimal risk to the privacy of individuals, based on, at least, the presence of the following elements:
- The requirements overlap but are not the same as those for waiver of consent and waiver of documentation of consent. There are additional requirements for HIPAA that are more stringent than for waiver under the Common Rule (research regulations).
- Even if documentation of informed consent can be altered or waived, HIPAA may still apply and authorization will need to be obtained if the requirements for a waiver of HIPAA are not met. In this situation, the investigator will need to obtain HIPAA authorization with a stand-alone HIPAA authorization form.
Active
Paper Records (e.g., consent forms, data files, medical records, etc.): Paper files related to human subjects participation in research must be securely stored on campus. Access to files should be restricted to key personnel and supervised by the principal investigator(s) of the study. Locked file cabinets ought to be used and preferably located in secured locations (i.e., locked office or laboratory). In the event that research activities are not carried on campus AND it is necessary to maintain the consent forms at the research site, copies of the signed consent forms should also be stored on a secure University location (either as a paper copy or in digital form).
IRBNet will send automatic reminders 60 days and 30 days before the expiration date, and an expiration alert the day an approval is set to expire. Please plan to submit your continuing renewal application with enough time to allow for its review and approval. No recruitment, data collection or private identifiable data analysis activities shall be carried on under an expired protocol.
If no continuing review application is submitted for review, projects will be closed after expiration. Further data collection and/or identifiable data analysis will require a new submission for IRB review and approval.
Closure and Retention
Records to be maintained include: copies of all research proposals reviewed, scientific evaluations (if any), consent documents, progress reports, reports of injuries to subjects and other unanticipated problems, and copies of all correspondences between the IRB and the investigator(s). Records may be preserved in hard-copy, electronic or other media form, and must be accessible for audit purposes. Records for completed projects should be stored in secure locations on campus with the same care used when the project was active.
If a researcher (faculty, staff or student) leaves UD, a copy of the research records must remain on campus. Students should coordinate storage of research records with their faculty advisor(s) and/or departments.
Destruction of Records
Researchers may retain de-identified data for future analysis in the context of the project for which the data were collected. Data are considered to be completely de-identified when ALL links between individual identity and the data are destroyed. Research data are not considered de-identified simply because names have been removed if they still contain information that might identify the participants such as date of birth, address, etc.
Questions
For further guidance, please contact the IRB Office at 302-831-2137.
Clinical Trial
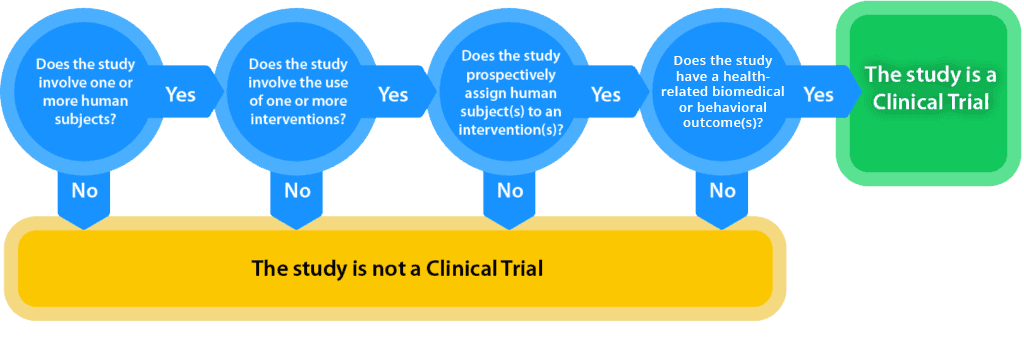
What is a Clinical Trial?
National Institutes of Health (NIH) generally defines a clinical trial as a research study in which one or more human subjects are prospectively assigned to one or more interventions (which may include placebo or other control) to evaluate the effects of those interventions on health-related biomedical or behavioral outcomes.
The definition of clinical trial used is slightly different depending on the requirement:
- Per FDAAA “Applicable Clinical Trials” generally include prospective interventional studies (with one or more arms) of FDA-regulated drugs, biological products or devicesthat meet one or more of the following conditions (regardless of funding source):
- The trial has one or more sites in the United States
- The trial is conducted under an FDA investigational new drug application or investigational device exemption
- The trial involves a drug, biologic or device that is manufactured in the United States or its territories and is exported for research
An interactive decision tool of the FDAAA applicable clinical trials definition is offered at https://grants.nih.gov/clinicaltrials_fdaaa/ACTs_under_FDAAA.htm enabling users to identify if the FDA requirements would apply to a specific study. - Per NIH, a Clinical Trial is a research study in which one or more human subjects are prospectively assigned to one or more interventions (which may include placebo or other control) to evaluate the effects of those interventions on health-related biomedical or behavioral outcomes.

- The ICMJE defines a clinical trial as any research project that prospectively assigns people or a group of people to an intervention, with or without concurrent comparison or control groups, to study the cause-and-effect relationship between a health-related intervention and a health outcome. Health-related interventions are those used to modify a biomedical or health-related outcome; examples include drugs, surgical procedures, devices, behavioral treatments, educational programs, dietary interventions, quality improvement interventions and process-of-care changes. Health outcomes are any biomedical or health-related measures obtained in patients or participants, including pharmacokinetic measures and adverse events.
For questions, send email to: clinicaltrials@udel.edu.
Registration is Required
Why is registration required?
Federal regulations and journal publications standards require that investigators register certain clinical studies in a publicly accessible database. www.ClinicalTrials.gov was created to support compliance with those requirements and standards. The diagram at right depicts the different requirements and how they, in some cases, may overlap.
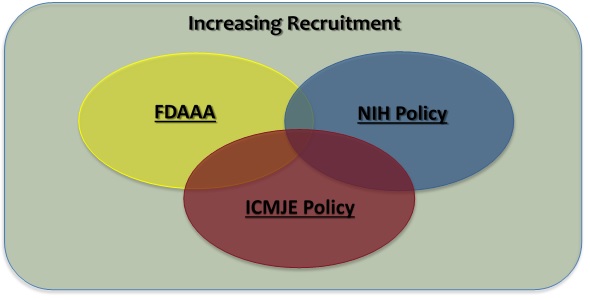
- The FDA Amendments Act of 2007 (FDAAA) requires “Applicable Clinical Trials” involving drugs, biological products and devices subject to the FDA regulations to be registered in ClinicalTrials.gov, regardless of the funding source for the study.
- The NIH Policy on the Dissemination of NIH-Funded Clinical Trial Information (“NIH Policy”) is complementary to the reporting requirements of FDAAA and establishes the expectation that all investigators conducting clinical trials funded, in whole or in part, by the NIH will ensure that these trials are registered and that results submitted to ClinicalTrials.gov.
- The International Committee of Medical Journal Editors (“ICMJE”) policy (adopted by over 1,000 journals) requires, and recommends that all medical journal editors require, registration of clinical trials in a public trials registry at or before the time of first patient enrollment as a condition of consideration for publication.
- In addition, an investigator may voluntarily decide to register a study not subject to any of the requirements above (e.g., an observational study with no intervention assignment and not intended to be published in a ICMJE journal) as a way to publicly advertise the research (i.e., recruitment of research subjects). ClinicalTrials.gov supports and encourages the registration of all research studies with human subjects, even if an explicit requirement does not apply.
For questions, send email to: clinicaltrials@udel.edu.
PI Information
I am the PI of a clinical trial. What do I do?
Once determined that a clinical trial will need to be registered in www.ClinicalTrials.gov the steps to follow are:
- If it is my first time registering a study: How do I obtain an account with ClinicalTrials.gov?Obtaining a username and password: If you do not have an account with ClinicalTrials.gov (i.e., no username and password), proceed to https://clinicaltrials.gov/:
- Click “Submit Studies” and then click “How to apply for an account.”
- Toward the bottom of the page, click the link, “PRS Administrator Contact Request Form.” Complete the contact request form for the appropriate organization (University of Delaware).
- You will receive an email from clinicaltrials.gov with the email address for the University of Delaware Protocol Registration System (PRS) Administrator account, clinicaltrials@udel.edu. Email the UD PRS Administrator account and request a username and password.
University of Delaware has a PRS Administrator account – do not create PRS “individual account” when registering with ClinicalTrials.gov. - If you do not know if you have an account or have forgotten your username, use the steps above to verify your username.
- How Do I Register a Trial?
- Prior to registration in ClinicalTrials.gov a UD research study must have received UD IRB approval. Pursuant to FDA Guidance, and NIH Policy, the following exact statement must be included in the informed consent documents of studies to be registered in ClinicalTrials.gov:
“A description of this clinical trial will be available on http://www.ClinicalTrials.gov, as required by U.S. Law. This Web site will not include information that can identify you. At most, the Web site will include a summary of the results. You can search this Web site at any time” - Once a username and password has been obtained, proceed to the ClinicalTrials.gov “PRS Login Page,” and enter the organization (UDelaware), username and password.
For basic help with using PRS, review the “Quick Start Guide” and the “PRS User’s Guide” found in the “Help” section of the PRS top main menu, which is accessible after logging with your username and password. - Complete all fields with information related to the trial. Once all of the information has been entered in the PRS record, marked as complete and released, an automated email will be sent to the UD PRS Administrator. Upon completion of the administrative review, which typically takes 3-5 business days, a UD PRS Administrator will approve and release the record to ClinicalTrials.gov, where the PRS team will review the record for quality control purposes prior to posting on the ClinicalTrials.gov website.
- Prior to registration in ClinicalTrials.gov a UD research study must have received UD IRB approval. Pursuant to FDA Guidance, and NIH Policy, the following exact statement must be included in the informed consent documents of studies to be registered in ClinicalTrials.gov:
Submit Results
How do I Submit Trial Results?
- Enter organization (UDelaware), your username, and your password on the “PRS Login Page.”
- Update the “Protocol Section” and release (submit) the record.
a. Ensure that the information in the Protocol Section is up-to-date before starting the Results Section (e.g., overall recruitment status, study start date, primary and study completion dates, actual enrollment, and arm and intervention information.)b. Begin results submission after the updated record has been published on ClinicalTrials.gov.
- Enter the required and optional results data elements. Scientific information is submitted as four separate modules: the modules allow for the entry and display of information in a series of data tables with supporting notes, but without narrative conclusions about the results. The scientific information instructions section provides detailed information on how to prepare the submission of each module and it includes links to instructional online presentations:
Participant Flow Summary of the progress of participants through each stage of a study, by study arm or comparison group. It includes the numbers of participants who started, completed and dropped out of each period of the study based on the sequence in which interventions were assigned. The module accommodates a wide range of study designs and allows for the description of key events following study enrollment but prior to group assignment. Baseline Characteristics Summary of the data collected at the beginning of the study for all participants, by study arm or comparison group. These data include demographics, such as age and gender, and study-specific measures (e.g., systolic blood pressure prior to exercise treatment). Outcome Measures and Statistical Analyses Summary of outcome measure values, by study arm or comparison group. It includes tables for each prespecified Primary Outcome and Secondary Outcome and may also include other prespecified outcomes, post hoc outcomes and appropriate statistical analyses. Adverse Events Summary of all anticipated and unanticipated serious adverse events and a tabular summary of anticipated and unanticipated other adverse events exceeding a specific frequency threshold. - Preview, inspect and release (submit) the record. For a description of criteria that should be addressed before releasing (submitting) the record, see “ClinicalTrials.gov Results Review Criteria.”
- When the record is released, an automated email will be sent to the UD PRS Administrator. Upon completion of the administrative review, which typically takes 3-5 business days, the UD PRS Administrator will approve and release the record to ClinicalTrials.gov, where a ClinicalTrials.gov staff member will review the record for quality control purposes prior to posting the results on the ClinicalTrials.gov website.
- Resources for the submission of results:
a. “Basic Results Data Element Definitions” contains descriptions of each required data item. b. Results data preparation checklists, simple results templates for each module, required data and a view of data elements in a tabular form are listed in the “Scientific Information” section of the “How to Submit Your Results” guidance on ClinicalTrials.gov. c. “Helpful Hints” contains tips on entering results data, including three examples of common study models (parallel design, crossover design and diagnostic accuracy studies) and measure types.
For questions, send email to the University of Delaware Research Office.
Good to Know
Additional Information
- There is a link at the bottom of each page on clinicaltrials.gov to submit a ticket for help: “CONTACT NLM HELP DESK.”
- ClinicalTrials.gov may be contacted with questions or for guidance via email at register@clinicaltrials.gov.
- If the question is about a specific study record, please provide the NCT Number or the Unique Protocol ID (if an NCT Number has not yet been assigned).
- Be detailed in your request. ClinicalTrials.gov generally responds to all emails within one business day.
Please see ClinicalTrials.Gov Guidance & Procedures here.
For questions, send email to: clinicaltrials@udel.edu.
ASSISTANCE
Compliance Hotline
Phone: (302) 831-2792
E: UD IRB Office
P: (302) 831-2137
F: (302) 831-2828
HUMAN SUBJECT LINKS
UD Policy & Procedure Manual
Involvement of Human Subjects in Research and Research-Related Activities
Belmont Report
Ethical principles for the protection of human subjects in research.
Common Rule
The Federal Policy for the Protection of Human Subjects or the “Common Rule” was published in 1991 and codified in separate regulations by 15 Federal departments and agencies, as listed here.
Office of Human Research Protections (OHRP)
United States Department of Health & Human Services Information
FDA Regulations relating to GCP and Clinical Trials